Dot plot
Category: other
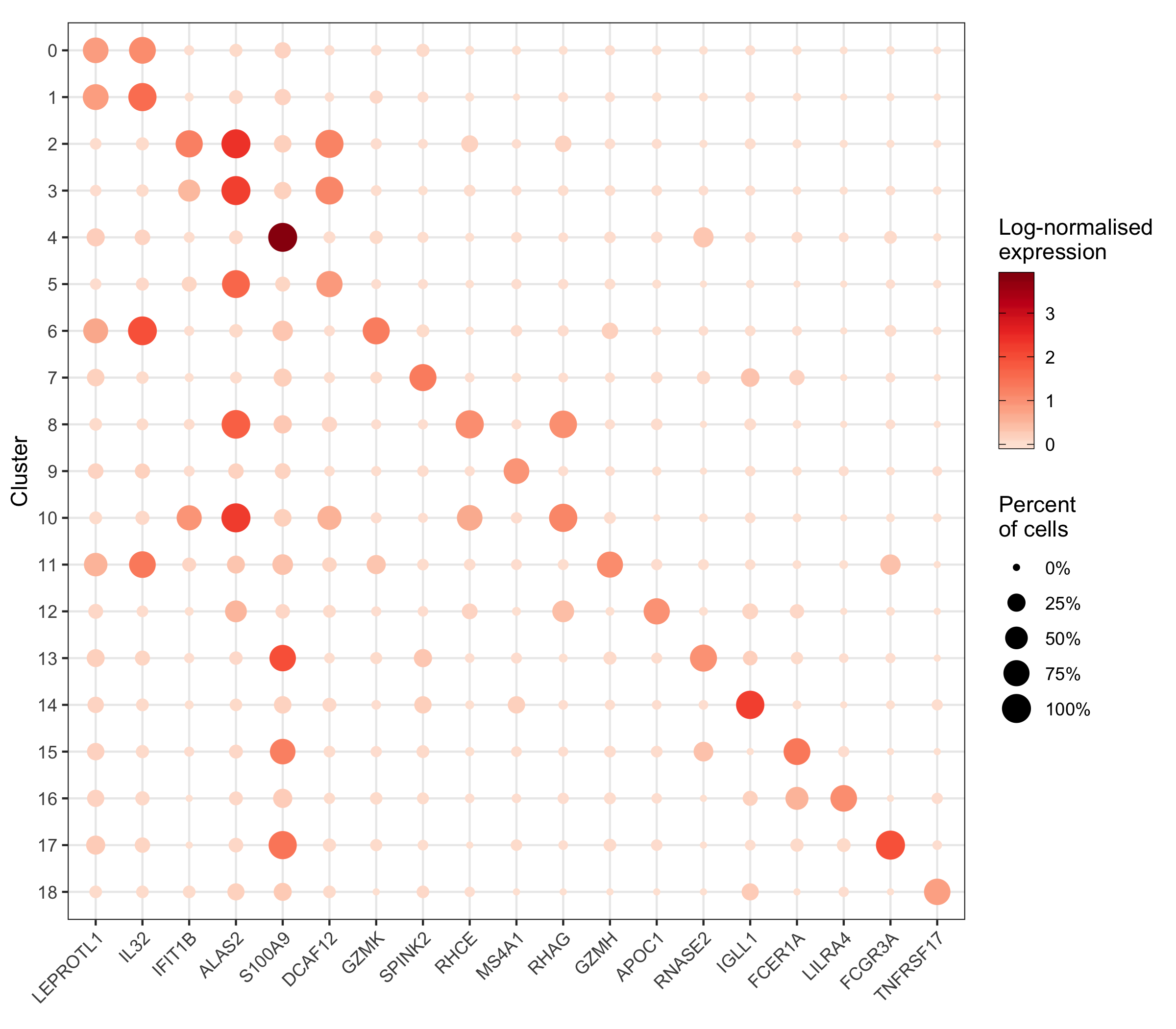
More context (and code) for this plot can be found in my scRNA-seq workflow in the chapter “Expression of individual genes”.
library(tidyverse)
library(Seurat)
# load a single cell expression data set (generated in the lab I work at)
seurat <- readRDS('seurat.rds')
# cells will be grouped by clusters that they have been assigned to
cluster_ids <- levels(seurat@meta.data$seurat_clusters)
# select a set of genes for which we want to show expression
genes_to_show <- seurat@misc$marker_genes$by_cluster %>%
group_by(cluster) %>%
arrange(p_val_adj) %>%
slice(1) %>%
pull(gene)
# for every cluster-gene combination, calculate the average expression across
# all cells and then transform the data into a data frame
expression_levels_per_cluster <- vapply(
cluster_ids, FUN.VALUE = numeric(length(cluster_ids)), function(x) {
cells_in_current_cluster <- which(seurat@meta.data$seurat_cluster == x)
Matrix::rowMeans(seurat@assays$SCT@data[genes_to_show,cells_in_current_cluster])
}
) %>%
t() %>%
as.data.frame() %>%
mutate(cluster = rownames(.)) %>%
select(cluster, everything()) %>%
pivot_longer(
cols = c(2:ncol(.)),
names_to = 'gene'
) %>%
rename(expression = value) %>%
mutate(id_to_merge = paste0(cluster, '_', gene))
# for every cluster-gene combination, calculate the percentage of cells in the
# respective group that has at least 1 transcript (this means we consider it
# as expressing the gene) and then transform the data into a data frame
percentage_of_cells_expressing_gene <- vapply(
cluster_ids, FUN.VALUE = numeric(length(cluster_ids)), function(x) {
cells_in_current_cluster <- which(seurat@meta.data$seurat_cluster == x)
Matrix::rowSums(seurat@assays$SCT@data[genes_to_show,cells_in_current_cluster] != 0)
}
) %>%
t() %>%
as.data.frame() %>%
mutate(cluster = rownames(.)) %>%
select(cluster, everything()) %>%
pivot_longer(
cols = c(2:ncol(.)),
names_to = 'gene'
) %>%
rename(cell_count = value) %>%
left_join(
.,
seurat@meta.data %>%
group_by(seurat_clusters) %>%
tally() %>%
rename(cluster = seurat_clusters),
by = 'cluster') %>%
mutate(
id_to_merge = paste0(cluster, '_', gene),
percent_cells = cell_count / n
)
# merge the two data frames created before and plot the data
p <- left_join(
expression_levels_per_cluster,
percentage_of_cells_expressing_gene %>% select(id_to_merge, percent_cells),
by = 'id_to_merge'
) %>%
mutate(cluster = factor(cluster, levels = rev(cluster_ids))) %>%
ggplot(aes(gene, cluster)) +
geom_point(aes(color = expression, size = percent_cells)) +
scale_color_distiller(
palette = 'Reds',
direction = 1,
name = 'Log-normalised\nexpression',
guide = guide_colorbar(frame.colour = "black", ticks.colour = "black")
) +
scale_size(name = 'Percent\nof cells', labels = scales::percent) +
labs(y = 'Cluster', color = 'Expression') +
coord_fixed() +
theme_bw() +
theme(
axis.title.x = element_blank(),
axis.text.x = element_text(angle = 45, hjust = 1)
)
ggsave('4.png', p, height = 7, width = 8)